模拟材料储氢效率的新方法
氢能有潜力成为实现联合国净零排放目标的关键措施,但其工业用途因其储存和处理困难而受到阻碍。氢气在极低的温度(-252°C)下会变成气体,这使得它在室温下的储存具有挑战性。氢与其储存材料之间的相互作用太弱,无法在室温下持续存在。这使得存储材料的设计对于实现将氢能引入日常使用的目标至关重要。
这是计算材料设计进来,很多的时间和精力氢燃料技术的发展过程中通过在计算机上设计的材料,并模拟其氢容量可以保存存储。但是这些预测在使用中变得非常有限,除非它们是准确的并且可以以合理的计算成本进行。在最近发表在ACSOmega上的一项研究中,科学家们开发了一种计算成本高但准确度高的预测储氢的新方法:“改进预测模拟的可靠性有助于加速氢燃料储存材料的开发,并导致一个更节能的社会,”领导这项研究的先进科学技术研究所(JAIST)的KentaHongo博士说。
物体之间的基本吸引力之一是范德华力,它根据原子或分子之间的距离定义了它们之间的相互作用。由于范德瓦尔斯力是相当复杂的量子过程的结果,传统的处理方法无法很好地描述它,因此到目前为止的模拟都处于对它的粗略估计的水平。但是在模拟储氢时这样做是否正确?这是本乡博士和团队最关心的问题。
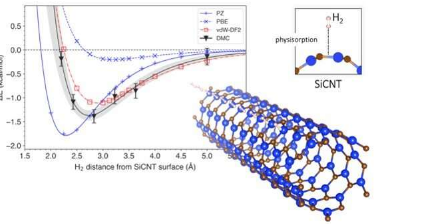
为了回答这个问题,他们研究了碳化硅纳米管,这是最有前途的储氢材料之一。使用称为扩散蒙特卡罗(DMC)的计算技术,他们创建了一个模型,该模型在模拟碳化硅纳米管中的氢存储时考虑了范德华力。大多数传统模型将氢和碳化硅纳米管之间的相互作用作为一个整体考虑,但DMC方法利用超级计算机的能力,通过跟踪单个电子的排列来忠实地重建相互作用机制。这使得DMC模型成为迄今为止最准确的预测方法。使用DMC模型,研究人员还能够预测将氢从其储存中取出需要多少能量,以及氢离碳化硅纳米管表面有多远。然后,他们将建模的结果与通过传统预测方法获得的结果进行了比较。
传统的预测方法通常基于称为密度泛函理论(DFT)的计算技术。DFT使用描述电子密度空间变化的泛函(量子相互作用的模型描述)来确定复杂系统的属性。虽然已经有几项基于DFT的研究关于碳化硅纳米管上的氢存储,但没有一项研究将范德华力纳入他们的预测中。然而,范德华修正的DFT泛函已被用于其他材料的预测。Hongo博士和团队使用范围广泛的DFT泛函模拟了储氢,这些泛函具有范德瓦尔斯修正,而那些没有。他们发现,没有范德华修正的DFT泛函将储氢所需的能量误估了4-14%。另一方面,范德瓦尔斯校正的DFT泛函产生的结果与DMC的结果非常相似。此外,他们发现范德华力对存储能量的贡献约为9-29%,这几乎是微不足道的。
Hongo博士认为,这些发现可以成为储氢模拟技术进一步创新的垫脚石。“虽然DMC方法的计算成本很高,但它可以用来阐明每种预测方法的特性(预测误差的趋势)。这将帮助我们了解应该信任哪个预测,以及如何修改预测方法以使其更有用,”他解释说。
标签: